Vol 3 | Issue 2 | May-Aug 2017 | page: 15-19 | B G Chenna Keshava, Padmini P Giri, Tejaswi Manubolu, J N Durga Rao Yadavalli
Authors: B G Chenna Keshava [1], Padmini P Giri [1], Tejaswi Manubolu [1], J N Durga Rao Yadavalli [2]
[1] Department of Critical Care, MS Ramaiah Hospital, Bengaluru, Karnataka, India.
[2] Department of General Medicine, MS Ramaiah Hospital, Bengaluru, Karnataka, India.
Address of Correspondence
Dr. B G Chenna Keshava,
Department of Critical Care, MS Ramaiah Hospital, Bengaluru – 560 054, Karnataka, India.
Phone: +91-9886051598. Tel.: +080-23492386.
E-mail: ckeshava7@gmail.com
Abstract
Introduction: Altered sensorium in adult ICU is very common and frequently multi-factorial. A systematic clinical examination and laboratory workup is quintessential in finding the cause. One amongst them is hyperammonemia. Hyperammonemia in adults is commonly associated with porto-systemic shunt with or without liver failure. In patients without liver failure or porto-systemic shunt, hyperammonemia can be caused by urea cycle enzyme deficiency. The features are of metabolic encephalopathy and lead to coma or death. Though they are rare the most commonly described enzyme deficiencies are ornithine transcarbamylase and carbamyl phosphate synthetase deficiency.
Case Report: A 35 year male patient was admitted with cough and intermittent fever was diagnosed to have pulmonary tuberculosis and was started on anti tubercular regimen (ATT). He was incoherent and drowsy in the second week of treatment. He had a very high ammonia with normal liver function test. A urea cycle enzyme deficiency targeted workup revealed CPS (carbamyl phosphate synthetase) deficiency which responded well to supportive therapy and was discharged home.
Conclusion: This case-study highlights the prevalence of urea cycle enzyme deficiency in adults. Though uncommon among adults it should form a part of differential diagnosis for hyperammonemia with normal liver function test. Though the diagnosis is expensive the treatment is quite affordable and patient compliance can be ensured with preventive measures. Timely assessment and laboratory backup can help in clinching the diagnosis and preventing long term complications.
Keywords: Carbamyl phosphate synthetase, hyperammonemia, ornithine transcarbamylase, urea cycle enzyme.
Introduction
Hyperammonemic coma due to urea cycle enzyme deficiency is commonly seen in children and rarely in adults. Hyperammonemia (HA) in the absence of hepatic failure is rare amongst adults. The most common causes are drug induced, enzyme deficiencies, portosystemic shunts, and urinary tract infections with urea-splitting organisms. Enzyme deficiency manifesting in elderly is not common. Altered sensorium in adults can be multifactorial. Herein, we describe a patient who developed altered sensorium after admission to hospital and was accidentally detected to have CPS-1 deficiency. He improved well on supportive care and was discharged home.
Case Report
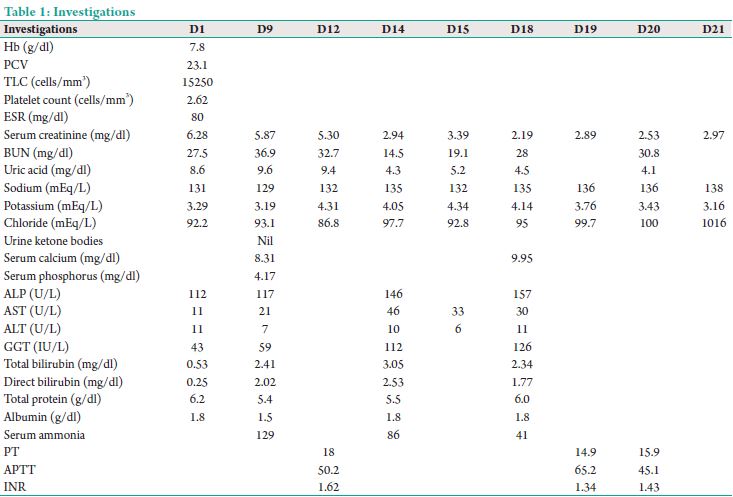
This was a 35-year-old male known case of chronic kidney disease (CKD) on conservative management/rheumatic heart disease/hypertension/anemia presented with intermittent fever and cough for 2 months. He was treated outside with a course of antibiotics before admission with no improvement and hence was referred to our center. On admission patient was drowsy barely responding to verbal commands and his vitals were blood pressure of 110/80 mmHg, heart rate of 100 bpm, temperature of 97°F, weight of 36 kg, and saturations of 97 on 6 L oxygen. The patient was admitted to intensive care unit (ICU) with suspicion of fever of unknown origin and septic workup initiated. Initial investigations as in Table 1.
His initial blood workup was essentially negative including a sputum for acid fast bacilli and Mantoux. Computed tomography chest showed a left lung cavity with a tree in bud appearance which was suggestive of tuberculosis or a vasculitis. The patient was started on anti-tubercular drugs (4 drugs) on day 5 and shifted out of ICU. Over the next 3-5 days, patient became afebrile and was planned for an arteriovenous fistula in view of the worsening renal functions. On D9 of admission noted to be drowsy and incoherent and was shifted back to ICU for further monitoring and care. In ICU noted to have a Glasgow coma scale (GCS) of 9/15 with severe flapping tremors and incoherent speech. Neuroimaging was essentially normal. He
was investigated for sepsis/dyselectrolytemia/antituberculosis therapy (ATT) induced hepatotoxicity/nephrotoxicity. His investigations showed an ammonia of 130 μmol/L (normal 20-36), serum sodium 120 mEq/L in the presence of normal liver function test (LFT). A glucometer random blood sugar, arterial blood gas (ABG), and urine ketones done at the same time were within normal limits. His blood urea was 36 and creatinine was 5.87. A workup for hyperammonemia (HA) was initiated. Serum citrulline level was <10 (normal 11-45), and urine orotic acid was absent. The overall findings suggested carbamoyl phosphate synthetase (CPS) deficiency. All hepatotoxic drugs were withheld, and alternate regimen of ethambutol and ofloxacin was initiated. He did improve with antiencephalopathy measures and sodium benzoate (3 g 3 times a day). He was in ICU for a period of 2 weeks, and during his stay, the patient was intubated and ventilated for 72 h for low GCS. He was successfully extubated after 3 days and supported by intermittent hemodialysis. Over a period of 2 weeks, he started making good urine, and his renal functions improved. His ammonia remained within normal limits with modified ATT regimen and sodium benzoate. His creatinine on shift out from ICU was 3.2. He was fully conscious oriented and afebrile at discharge.

Discussion
Acute hyperammonemic (HA) encephalopathy has significant morbidity and mortality unless treated on time. HA in adults without hepatic dysfunction or non-hepatic HA coma/encephalopathy (NHHC) is an unusual phenomenon [1]. Ammonia in the body is a byproduct of protein metabolism. It freely crosses blood–brain barrier and at significant concentrations is toxic. Plasma concentration of ammonia is maintained by a complex integrated mechanism involving enterohepatic circulation and the urea cycle, which detoxifies and converts ammonia to urea which is electrically neutral and soluble in water. When compared to ammonia which raises the cell pH when it accumulates in a cell, urea is a relatively safe vehicle to transport and excrete nitrogen As a result of this complicated mechanism ammonia remains low (<40 mmol/L) except when the urea cycle cannot handle the ammonia load. The most common cause in adults is liver damage. The most common etiologies of NHHC (Table 2) are congenital enzyme defects, drug induced states (which act on urea cycle), portosystemic shunts, and urinary tract infections with urea-splitting organisms. Since they are less common and more likely to be missed leads to diagnosis delayed with disastrous consequences. There is an effective treatment for most of them but should be started early to prevent long-term neurological damage and early death [2].
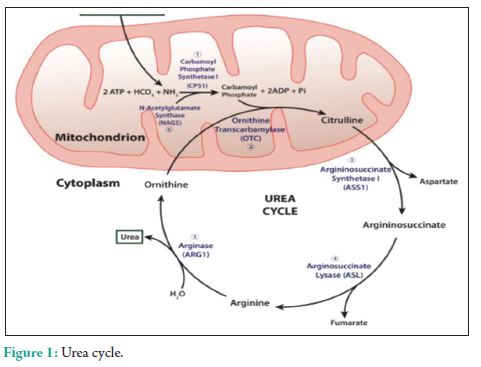
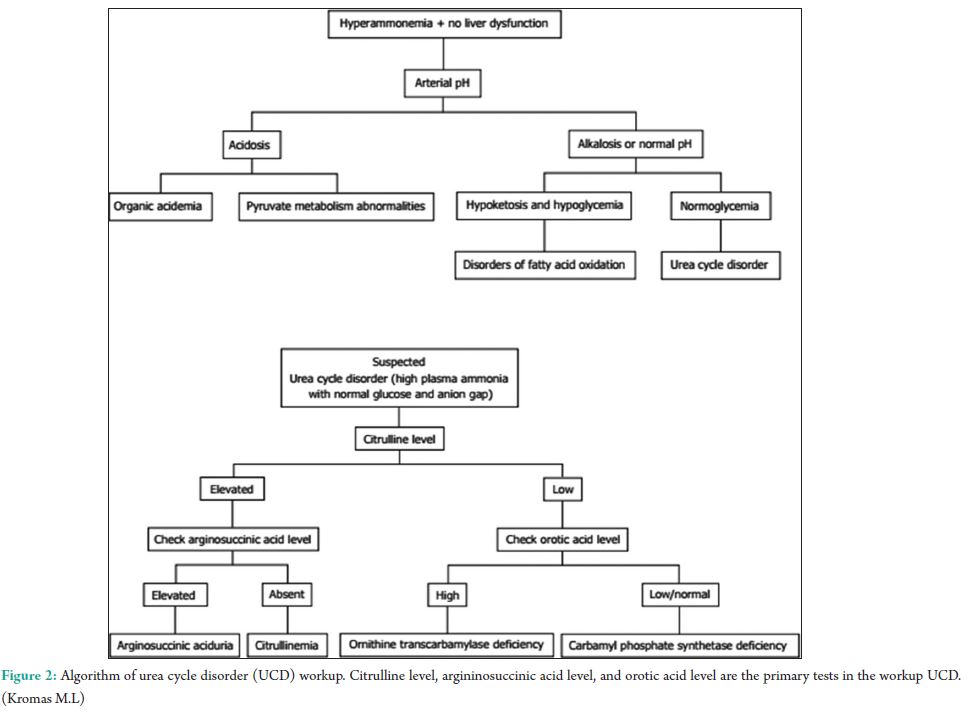
The urea cycle (Fig. 1) in hepatocytes is mediated by 6 enzymes (N-acetyl-glutamate synthetase, CPS, ornithine transcarbamylase (OTC), argininosuccinate synthetase, argininosuccinate lyase, and arginase. Deficiencies in any of these enzymes are collectively referred to as the urea cycle disorders (UCDs), which are inherited [3]. Complete absence or partial inactivity of these enzymes can predispose patients to episodic, life-threatening HA. Acute HA manifests as anorexia, vomiting, and altered mental status. Lethargy, progressing to coma, and possibly death if any delay in the treatment. Seizures, ataxia, asterixis, slurred speech, tremors, weakness, muscle tone abnormalities, and hypothermia are other common manifestations. Individuals with partial enzyme deficiency typically manifest outside the neonatal period. Clinical features may be subtle and milder, in such late-onset cases, leading to delayed diagnosis with devastating neurological sequelae and death. Psychiatric symptoms in late-onset UCDs, including hyperactive behavior, mood disturbances, self-injurious behavior, and psychosis, are noted [4]. A tendency to avoid dietary protein is common [5]. Acute HA affects the brain white matter selectively and causes astrocyte swelling and global cerebral edema. Changes involving the deep insular and perirolandic sulci may be reversible. However, in cases of severe HA, permanent changes occur. Patients with late-onset disease were more likely to survive an episode of HA than neonates (98% vs. 73%, P = 0.001). Dialysis was used in conjunction with alternative pathway therapy in 60% of neonatal and 7% of late-onset episodes [6]. The diagnosis of UCD is dependent on biochemical distinguishing features. The routine investigations in UCD patient would reveal a normal or slightly deranged LFT with a markedly elevated serum ammonia. In neonates, ammonia levels are ≥1000 mmol/L, whereas other etiologies seldom present with ammonia levels <200-300 mmol/L. In addition, UCD is suggested by normal blood glucose and anion gap, as well as respiratory alkalosis from central hyperventilation. The initial workup of a UCD should include glucose, electrolytes, amino acids, serum ammonia, lactate, urine organic acids, orotic acid and ABGs looking for arterial pH, and carbon dioxide. Gene mutations are not revealed in DNA analysis in up to 20% of patients with OTC deficiency. Therefore, other lab measures may give an indication as to the etiology of HA without a history of the liver disease [7]. A systematic workup to identify UCD is depicted in Fig. 2. Patients with CKD with high urea might require due consideration, as ammonia is a precursor for urea. A case-control study to assess the serum levels of ammonia in uremic encephalopathy patients in comparison to non-uremic patients did not show any significant difference [8]. Our patient did not have very high urea to attribute. The causes of HA in the presence of normal liver functions are not many. In our patient, the altered level of consciousness could have been multifactorial, which included hyponatremia, HA, and probably uremia. The very high levels of ammonia (130 mmol/L) with a near normal LFT prompted us to consider the workup for urea cycle enzymes and significant logistical support helped us to narrow down the cause to CPS-1 deficiency, probably a partial deficiency. The anti-tubercular regimen could have worsened his CPS-1 deficiency [9]. The multiple comorbidities, an early and adequate clinical response, made us think twice regarding liver biopsy and enzyme assay. A DNA analysis was planned for a later date, but the patient was lost to follow-up.
Conclusion
An altered level of consciousness though has multiple causes; an elevated ammonia (nearing 200 mmol/L) with normal LFT should prompt a workup for UCD. UCD though a rare entity, a high index of suspicion in the appropriate settings should clinch the diagnosis. Although the workup is tedious, the definitive management, which includes is sodium phenyl acetate and sodium benzoate, in conjunction with other therapies, such as intravenous arginine hydrochloride, hemodialysis and the provision of adequate calories to prevent catabolism, effectively lower plasma ammonium levels, and result in survival in the majority. The positive side is, sodium benzoate is extremely cheap, and the patient response is quick and rewarding.
References
1. Chenna Keshava BG, Durga Rao Yadavalli JN, Giri M, Rao R. Non-hepatic hyperammonemic coma: A case report.
Asian J Pharm Clin Res 2016;9:2-4.
2. Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest 2007;132:1368-78.
3. Ah Mew N, Lanpher BC, Gropman A, Chapman KA, Simpson KL, Summar M, et al. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, et al., editors. Urea cycle disorders consortium. Urea Cycle Disorders Overview, GeneReviews®; 2003 April, 29. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1217. Last updated on 2015 Apr 09].
4. Enns GM. Neurologic damage and neurocognitive dysfunction in urea cycle disorders. Semin Pediatr Neurol
2008;15:132-9.
5. Gropman AL, Summar M, Leonard JV. Neurological implications of urea cycle disorders. J Inherit Metab Dis
2007;30:865-79.
6. Enns GM, Berry SA, Berry GT, Rhead WJ, Brusilow SW, Hamosh A. Survival after treatment with phenylacetate
and benzoate for urea-cycle disorders. N Engl J Med 2007;356:2282-92.
7. Yamaguchi S, Brailey LL, Morizono H, Bale AE, Tuchman M. Mutations and polymorphisms in the human ornithine. transcarbamylase (OTC) gene. Hum Mutat 2006;27:626-32.
8. Jothimalara SS Significance of blood ammonia in uremic encephalopathy. Sri Ramachandra J Med 2009;1(1):14-6.
9. Kromas ML, Mousa OY, John S. Hyperammonemia-induced encephalopathy: A rare devastating complication of bariatric surgery. World J Hepatol 2015 8;7:1007-11.
| How to Cite this Article: Keshava BGC, Giri PP, Manubolu T, Yadavalli JNDR. A rare and unusual presentation of urea cycle enzyme deficiency in critically ill adults: – Case report. Journal of Anaesthesia and Critical Care Case Reports May-Aug 2017;3(3):15-19. |
